Generative flows on discrete state-spaces: Enabling multimodal flows with applications to protein co-design
Summary
Technical Details
Discrete Flow Matching
For each real datapoint $x_1$, we define a very simple two-state interpolation between the mask token $M$ and the clean value $x_1$:
\[p_{t|1}^{\text{mask}}(x_t \mid x_1) = t \, \delta\{x_t, x_1\} + (1 - t) \, \delta\{x_t, M\}, \quad t \in [0, 1]\]Because $p_{t \mid 1}$ is analytically available, the unconditional flow is just:
\[p_t(x_t) = \mathbb{E}_{x_1 \sim p_{\text{data}}} \left[ p_{t \mid 1}(x_t \mid x_1) \right]\]Training the denoiser
The goal is a network $\hat{p}^{\theta}_{1 \mid t}(x_1 \mid x_t)$ that predicts the clean value.
Because $p_{t \mid 1}$ is in closed form, we can generate training pairs $(x_t, x_1, t)$ without simulating the CTMC.
Loss (cross-entropy)
\[\mathcal{L}_{\text{CE}}(\theta) = \mathbb{E}_{\substack{ x_1 \sim p_{\text{data}} \\ t \sim \mathcal{U}(0,1) \\ x_t \sim p_{t|1}(x_t \mid x_1) }} \left[ -\log \hat{p}^{\theta}_{1|t}(x_1 \mid x_t) \right]\]Key fact: $\mathcal{L}_{\text{CE}}$ does not depend on the rate matrix you will choose for inference, so training and sampling can be treated separately.
Constructing a time‑dependent rate matrix
During inference, we simulate a CTMC whose marginal at every time $t$ is $p_{t\mid 1}(x_t \mid x_1)$.
One valid choice (there are infinitely many) is the minimal-jump matrix $R_t^{\star}$.
Minimal-jump matrix $R_t^{\star}$
For the masked schedule the formula collapses to:
\[R_t^{\star}(x_t, j \mid x_1) = \frac{\delta\{x_t, M\} \, \delta\{j, x_1\}}{1 - t}, \quad 0 \leq t < 1\]Interpretation:
- You jump only when you are currently masked.
- You jump only to the correct clean value $x_1$.
- The hazard $(1 - t)^{-1}$ grows as $t \to 1$, guaranteeing that the jump occurs before time 1 with probability 1.
Multimodal Flows
Following FrameFlow, we refer to the protein structure as the backbone atomic coordinates of each residue.
The structure is represented as elements of $\mathrm{SE}(3)$ to capture the rigidity of the local frames along the backbone.
A protein of length $D$ residues can then be represented as:
\[\left\{ (x^d, r^d, a^d) \right\}_{d=1}^{D}\]where:
- $x \in \mathbb{R}^3$ is the translation of the residue’s Carbon-$\alpha$ atom,
- $r \in \mathrm{SO}(3)$ is a rotation matrix of the residue’s local frame with respect to the global reference frame, and
- $a \in {1, \ldots, 20} \cup {M}$ is one of 20 amino acids or the mask state $M$.
For brevity, we refer to the residue state as:
\[T^d = (x^d, r^d, a^d)\]and let the full protein’s structure and sequence be:
\[\mathbf{T} = \{ T^d \}_{d=1}^{D}\]We define the multimodal conditional flow as $p_{t\mid 1}(\mathbf{T}_t \mid \mathbf{T}_1)$ to factorize over both dimensions and modality.
We impose independence over both residues and modalities:
\[p_{t\mid 1}(\mathbf{T}_t \mid \mathbf{T}_1) = \prod_{d=1}^{D} p_{t\mid 1}(x_t^d \mid x_1^d) \, p_{t\mid 1}(r_t^d \mid r_1^d) \, p_{t\mid 1}(a_t^d \mid a_1^d)\]Continuous schedules (identical to FrameFlow / Yim et al. 2023a):
-
Translation:
\[x_t^d = t x_1^d + (1 - t)x_0^d, \quad x_0^d \sim \mathcal{N}(0, I)\] -
Rotation: \(r_t^d = \exp_{r_0^d} \left( t \log_{r_0^d}(r_1^d) \right), \quad r_0^d \sim \mathcal{U}_{\mathrm{SO}(3)}\)
Discrete mask schedule (our focus):
\[p_{t\mid 1}(a_t^d \mid a_1^d) = t \, \delta\{a_t^d, a_1^d\} + (1 - t) \, \delta\{a_t^d, M\}\]Trajectory generator
We build a single multimodal process whose marginal at every time matches the factorised $p_{t\mid 1}$.
- Continuous modalities. Choose velocity fields that individually produce the conditional marginals:
With Euler integration (step $\Delta t$):
\[x_{t + \Delta t}^d = x_t^d + v_x^d \Delta t, \quad r_{t + \Delta t}^d = \exp_{r_t^d}(\Delta t \, v_r^d)\]- Discrete modality — minimal-jump CTMC
Training objective
Sample $t \sim \mathcal{U}(0, 1)$ and corrupt $\mathbf{T}_1$:
\[\mathbf{T}_t = \left\{ t x_1^d + (1 - t) x_0^d,\ \exp_{r_0^d}\left[t \log_{r_0^d}(r_1^d)\right],\ \text{Bernoulli}(t, a_1^d, M) \right\}_{d=1}^{D}\]The network predicts:
\[\hat{x}_1^d,\ \hat{r}_1^d,\ \hat{\pi}(\cdot \mid \mathbf{T}_t, t)\]Independent losses per modality:
\[\mathcal{L}(\theta) = \mathbb{E} \left[ \underbrace{\frac{\|\hat{x}_1^d - x_1^d\|_2^2}{1 - t}}_{\text{trans.}} + \underbrace{\frac{\| \log_{r_t^d}(r_1^d) - \log_{r_t^d}(r^d) \|_2^2}{1 - t}}_{\text{rot.}} - \underbrace{\log \hat{\pi}_{a_1^d}}_{\text{amino acid}} \right]\]To enable flexible sampling options, we can use a noise level for the structure, t, that is independent to the noise level of the sequence, $\tilde{t}$.
Architecture
The paper uses an IPA backbone from FrameFlow, plus a transformer head for sequence logits; any SE(3)-equivariant encoder-decoder that outputs these three targets suffices.
Sampling algorithm (Euler + CTMC-thinning)
input : trained network f_θ , step Δt (e.g. 1e-4)
output: protein sample 𝒯 ∼ p_data
# 0. initialise at pure noise
t ← 0
for d = 1 … D:
x^d ← 𝒩(0, I) # translation noise
r^d ← Uniform_SO(3) # rotation noise
a^d ← M # fully masked sequence
# 1. forward simulation
while t < 1:
# 1.1 network predictions
{x̂_1^d, r̂_1^d, π̂^d} ← f_θ({x^d, r^d, a^d}, t)
# 1.2 continuous Euler step
x^d ← x^d + (x̂_1^d − x^d)/(1 − t) * Δt
r^d ← Exp_{r^d}( (log_{r^d}(r̂_1^d))/(1 − t) * Δt )
# 1.3 sequence CTMC step (per residue)
if a^d == M:
λ ← 1 / (1 − t) # total rate
stay_prob ← max(0, 1 − λ * Δt) # Poisson thinning
if Uniform(0,1) > stay_prob: # jump occurs
a^d ← Cat(π̂^d) # choose new residue
t ← t + Δt
return {x^d, r^d, a^d}_{d=1}^D
Notes
- 1.3 uses the minimal-jump rule. To introduce extra randomness, add an $\eta$-scaled detailed-balance matrix before the thinning step.
- $\Delta t$ can be made adaptive (e.g., $\Delta t \propto 1 - t$) to cope with the diverging rate near $t = 1$.
- You may stop at $t = 0.999$ and set any remaining $M$ tokens to $\arg\max \hat{\pi}$.
Experiments
Training
Training data consisted of length 60-384 proteins from the Protein Data Bank (PDB) (Berman et al., 2000) that were curated in Yim et al. (2023b) for a total of 18684 proteins.
Distillation
Our analysis revealed the original PDB sequences achieved worse designability than PMPNN. We sought to improve performance by distilling knowledge from other models.
- we first replaced the original sequence of each structure in the training dataset with the lowest scRMSD sequence out of 8 generated by PMPNN conditioned on the structure.
- we generated synthetic structures of random lengths between 60-384 using an initial Multiflow model and added those that passed PMPNN 8 designability into the training dataset with the lowest scRMSD PMPNN sequence.
Metrics
-
Designability. The generated structure is called designable if scRMSD <2˚ A. Designability is the percentage of designable samples.
-
Diversity. We use FoldSeek to report diversity as the number of unique clusters across designable samples.
-
Novelty. Novelty is the average TM-score of each designable sample to its most similar protein in PDB.
Co-design results

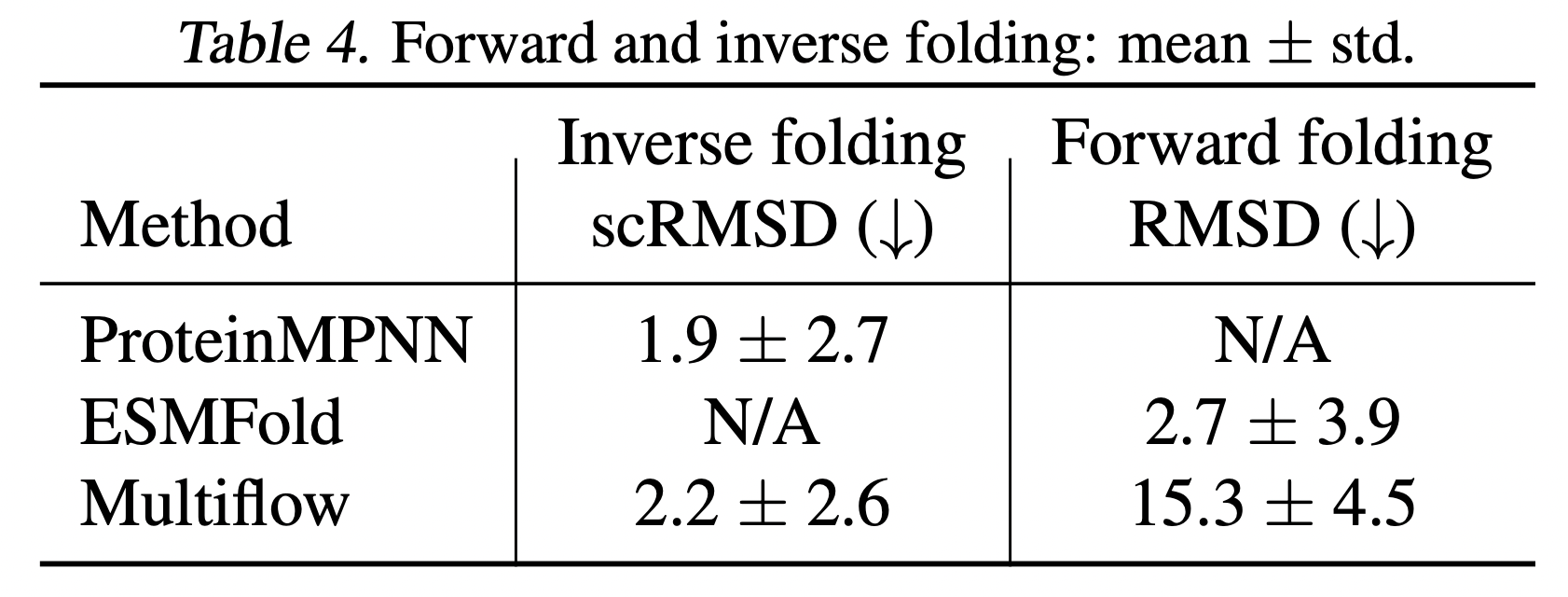
Forward and inverse folding